
Comment le vieillissement influence-t-il le système immunitaire pour aboutir à cette inflammation froide, à bas bruit, responsable des multiples pathologies associées au vieillissement que les anglo-saxons nomment “inflammaging”?
Le système immunitaire inné identifie les dangers de toutes sortes grâce à des récepteurs innés qui différencient le soi du non soi[1]. Les macrophages sont au centre de l’”inflammaging”[2][3], au sein de multiples tissus et organes, tels que le microbiote. Lors du vieillissement, l’activité des macrophages évolue de la phagocytose comme activité dominante vers une activité pro-inflammatoire, à l’origine de la production accrue d’interleukine 6 (IL-6), d’IL-1β et de Tumor Nerosis Factor (TNF[4]). Par ailleurs, tant les précurseurs que les macrophages eux-mêmes voient leur nombre diminuer dans la moëlle osseuse.
D’un point de vue fonctionnel, plusieurs types d’agents, les germes microbiens, les débris cellulaires, les nutriments et le microbiote, interagissent avec des récepteurs intra-cellulaires qui déclenchent la réponse immunitaire innée. Les molécules endogènes sont la source principale de cette inflammation. Altérées, déplacées, issues de débris cellulaires ou de cellules dysfonctionnelles, elles sont reconnues par les récepteurs du système immunitaire inné. Les récepteurs du cytosol des macrophages jouent un rôle important dans la formation de complexes protéiques, les inflammasomes.
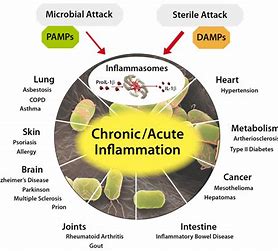
L’inflammasome est un groupe de récepteurs du cytosol qui contrôlent la production de cytokines pro-inflammatoires comme IL-β et IL-18, une cytokine cruciale dans la défense immunitaire et l’inflammation. C’est un complexe multiprotéique qui régule diverses fonctions physiologiques comme la pyroptose[5] et la réparation tissulaire. Le complexe NLRP3 notamment, est stimulé par le potassium extra-cellulaire, les espèces réactives de l’oxygène issues des mitochondries, les protéases lysosomales.[6]
Le récepteur NLRP3 conduit à la formation de l’inflammasome NLRP3, le plus étudié à ce jour. Le récepteur réagit à la présence de microbes (virus, bactéries, champignons), de toxines, d’agents irritants ( silice, aluminium, UV), de cristaux d’urate de Na, de B-amyloid, d’agrégats protéiques[7], ou d’ATP extra-cellulaire provenant de cellules tumorales.

L’induction des inflammasomes s’accompagne de la formation d’autophagosomes qui limitent la portée de la réaction inflammatoire et permettent aux cellules de se débarasser des pyroptosomes[8], ces composants inflammatoires qui conduisent à la destruction de la membrane cellulaire et à la libération intensive de cytokines[9].
Nous avons vu dans les précédents chapitres comment la diminution de l’autophagie conduisait à l’agrégation de protéines, et à l’accumulation de mitochondries déficientes. Nous avons établi le rôle des mitochondries dans la dégradation de cette maintenance cellulaire, par la production d’espèces réactives de l’oxygène et le stress oxydatif[10]. Les mitochondries sont la source principale de espèces réactives de l’oxygène requises pour l’activation d’inflammasomes[11].
Les mitochondries jouent un rôle important dans la régulation de la réponse immunitaire innée. Elles sont impliquées dans l’activation de l’inflammasome NLRP3 en présence des espèces réactives de l’oxygène, lorsqu’elles sont déficientes en protéines de l’autophagie[12]. Or les mitochondries sont affectées par la production élevée d’espèces réactives de l’oxygène, à la fois directement et indirectement, du fait des mutations induites par celles-ci sur l’ADN mitochondrial[13].
Ces espèces réactives de l’oxygène stimulent les inflammasomes, les plateformes intra-cellulaires protéiques réactives à la perception d’un signal de danger, comme le stress oxydatif. Ces inflammasomes activent des cytokines qui provoquent une réponse inflammatoire. Celle-ci accélère le vieillissement en inhibant davantage l’autophagie[14].
L’inflammation en retour réprime la capacité autophagique, et stimule l’inflammasome via les glucocorticoïdes qui provoquent l’expression de NLRP3 dans les macrophages. Un taux de cortisol élevé pourrait ainsi paradoxalement augmenter la réponse inflammatoire de l’organisme[15].
Le déclin de l’autophagie altère le bon fonctionnement cellulaire et expose les cellules à l’activation de l’inflammasome. L’expression progressivement croissante au fil du temps des marqueurs de l’inflammation produit une stimulation prolongée du système immunitaire.

Les dérèglements de l’autophagie perturbent le système immunitaire. L’inflammation globale de l’organisme stimule l’axe hypothalamo-hypophyso-surrénalien et augmente la sécrétion de cortisol, qui en retour, produit un hypercatabolisme des protéines, notamment dans les muscles et les os[16], aboutissant à la sarcopénie. Elle provoque le développement d’une résistance à l’insuline et des troubles dans la régulation de l’énergie cellulaire.
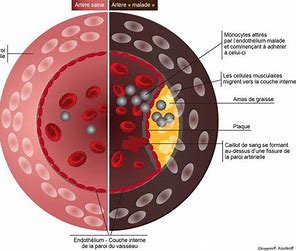
La dysrégulation de l’ inflammasome se manifeste par une vulnérabilité accrue aux infections et est associée à diverses pathologies liées à une inflammation chronique, comme les pathologies auto-immunes., le lupus erythémateux, la maladie de Crohn[17], l’obésité, le diabète de type 2, l’athérosclérose[18], et les cancers[19]. La capacité autophagique est également compromise dans certaines pathologies neuro-dégénératives, comme Alzheimer[20], et Parkinson[21].
La “metaflammation” est ainsi une inflammation chronique d’origine métabolique, médiée par les inflammasomes. Ce processus auto-immun accélère le vieillissement et provoque l’aggravation des maladies chroniques qui accélèrent à leur tour le processus de sénescence[22].
Les pathologies associées au vieillissement correspondent à cet état avancé de dysrégulation de l’autophagie, ayant conduit à une accumulation de déchets et à une réaction inflammatoire chronique d’origine métabolique, processus auto-entretenu qui aboutit au déclin de l’organisme.

Il existe des solutions comportementales, nutritionnelles, pharmacologiques, qui permettent de ralentir ce processus et de préserver les fonctions cellulaires, pour “âger” en bonne santé. A suivre…
Pour en savoir plus, notre ouvrage sur l’avenir du vieillissement: “Vieillir, un destin remédiable“.
[1] Akira S. Innate immunity and adjuvants. Philos Trans R Soc Lond B Biol Sci. 2011;366:2748–2755.
[2] Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S. Inflammaging and ‘Garb-aging’. Trends Endocrinol Metab. 2017 Mar;28(3):199-212. doi: 10.1016/j.tem.2016.09.005. Epub 2016 Oct 24. PMID: 27789101.
[3] Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. NY. Acad. Sci. 2000;908:244–254.
[4] Shaw AC, Joshi S, Greenwood H, Panda A, Lord JM. Aging of the innate immune system. Current Opinion in Immunology. 2010;22(4):507–513.
[6] Rathinam VAK, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nature Immunology. 2012;13(4):333–342.
[7] De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011;32:373–379.
[8] (PDF) The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation (researchgate.net)
[9] Suzuki T, et al. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 2007;3:e111
[10] Salminen A, Kaarniranta K, Kauppinen A. Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging (Albany NY). 2012 Mar;4(3):166-75. doi: 10.18632/aging.100444. PMID: 22411934; PMCID: PMC3348477.
[11] Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230.
[12] Nakahira K, Haspel JA, Rathinam VAK, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AMK. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011;8:222–230.
[13] Ozawa T. Genetic and functional changes in mitochondria associated with aging. Physiol Rev. 1997;77:425–464.
[14] Chen G, Shaw MH, Kim YG, Nunez G. NOD-like receptors: role in innate immunity and inflammatory disease. Annu. Rev. Pathol. Mech. Dis. 2009;4:365–398.
[15] Busillo JM, Azzam KM, Cidlowski JA. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. J. Biol. Chem. 2011;286:38703–38713.
[16] Giunta G. Exploring the complex relations between inflammation and aging (inflamm-aging): anti-inflamm-aging remodelling of inflamm-aging, from robustness to frailty. Inflamm. Res. 2008;57:558–563.
[17] Kuballa P, Huett A, Rioux JD, Daly MJ, Xavier RJ. Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS ONE. 2008;3:e3391.
[18] Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Aging-induced proinflammatory shift in cytokine expression profile in rat coronary arteries. FASEB J. 2003. 17:1183–1185.
[19] Yang C-S, Shin D-M, Jo E-K. The role of NLR-related protein 3 inflammasome in host defense and inflammatory diseases. International Neurourology Journal. 2012;16(1):2–12.
[20] Nixon RA, Yang DS. Autophagy failure in Alzheimer’s disease – locating the primary defect. Neurobiol. Dis. 2011;43:38–45.
[21] Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6:352–361.
[22] Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 2018 Oct;14(10):576-590. doi: 10.1038/s41574-018-0059-4. PMID: 30046148.