La dégradation du recyclage des mitochondries, c’est l’engrenage du vieillissement
Les mitochondries sont les centrales énergétiques cellulaires, qui se reproduisent à partir d’autres mitochondries, par fusion de plusieurs mitochondries qui deviennent géantes et division en mitochondries filles, selon les besoins de la cellule qui les abrite. Une cellule active peut produire 10 fois plus de mitochondries qu’une cellule quiescente. Le recyclage des mitochondries est une tâche majeure du dispositif de maintenance cellulaire, particulièrement chez les cellules à durée de vie longue, comme les neurones, les cellules myocardiques, les cellules des muscles squelettiques.
Dès son plus jeune âge, un organisme produit des mitochondries qui arrivent en fin de vie, d’abord en nombre limité, puis en augmentation avec l’âge. Nous allons voir que la difficulté croissante pour l’organisme d’éliminer et de recycler les mitochondries dysfonctionnelles installe la cellule dans un cercle vicieux, où l’accumulation d’organites défectueux altère progressivement le dispositif en charge de sa maintenance et de son recyclage: l’autophagie, ici la mitophagie plus précisément.
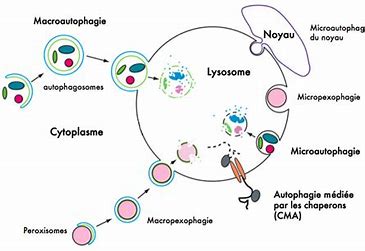
D’un point de vue fonctionnel, la mitophagie correspond à la formation de vacuoles autophagiques qui enrobent les éléments à recycler, et les relarguent dans le lysosome, une bulle faite de vacuoles qui fusionnent et se divisent constamment, reçoivent des enzymes et des substrats issus de la cellule ou provenant de l’extérieur. Après la dégradation des substrats dans le lysosome, les produits diffusent ou sont transportés activement au cytosol pour y être recyclés[1].
Nous avons vu dans le chapitre précédent que l’autophagie ralentissait progressivement; le nombre de mitochondries dysfonctionnelles augmente en corrélation avec ce ralentissement. Par voie de conséquence, la maintenance de l’équilibre protéique se dégrade. La durée de vie prolongée de mitochondries défectueuses, dûe au ralentissement de l’autophagie, augmente la perméabilité des membranes mitochondriales sous l’effet des espèces réactives de l’oxygène produites en plus grand nombre.
L’autophagie des mitochondries enrichit le lysosome en fer, ce qui le rend sensible au stress oxydatif. C’est l’évolution de la mitophagie caractéristique du vieillissement. Les mitochondries géantes ne sont plus accessibles à l’autophagie, et n’échangent plus leur contenu avec les mitochondries normales, selon un mécanisme de fusion habituel[2]. Les résidus mitochondriaux riches en fer[3] produisent la lipofuscine, qui fragilise la membrane lysosomiale, rendue ainsi sensible à l’oxydation. L’accumulation de composants cellulaires défectueux comme la lipofuscine résulte de la maintenance insuffisante de la protéostasie[4],
La lipofuscine est un pigment non dégradable qui s’accumule dans les cellules âgées, à faible taux de renouvellement, comme les neurones, les cellules cardiaques. C’est un polymère proche du plastique. Sa formation est le reflet de l’atteinte radicalaire au niveau du lysosome. La genèse de la lipofuscine est très proche de la formation des produits finaux de la glycation (AGEs) qui produisent rides, cataracte et rigidité des vaisseaux sanguins. Les sucres ne sont pas impliqués dans la formation de lipofuscine, formée de résidus protéiques oxydés associés au fer. Dans les lysosomes riches en lipofuscine se produisent des réactions de Fenton[5].
Les radicaux hydroxyles produits par les mitochondries géantes conduisent à la formation de lipofuscine et à la perméabilisation de la membrane lysosomale. Les mitochondries défectueuses s’accumulent encore plus maintenant que les lysosomes chargés en lipofuscine consomment leurs hydrolases, rendues inutilisables pour l’autophagie, et notamment la mitophagie. L’agglomération de lipofuscine dans le lysosome compromet la capacité de dégradation par autophagie, ce qui prolonge la durée de vie de protéines et d’organites dysfonctionnels, comme des mitochondries produisant toujours plus d’espèces réactives de l’oxygène qui à leur tour contribuent à la genèse de la lipofuscine et à l’apoptose cellulaire. Le cercle vicieux du vieillissement se met en place. Les mitochondries senescentes et les lysosomes chargés en lipofuscine remplacent progressivement les organites normaux.
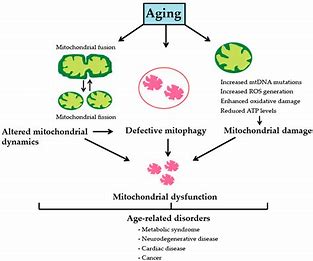
L’accumulation de composants cellulaires défectueux comme la lipofuscine résulte de cette maintenance insuffisante de la protéostasie[6], tandis que l’autophagie diminue et que le nombre de mitochondries dysfonctionnelles augmente. La lipofuscine altère la fonction du lysosome, qui n’élimine plus les mitochondries déficientes, ce qui conduit à l’augmentation de la production des espèces réactives de l’oxygène et à la stimulation de l’inflammasome[7], que nous aborderons au prochain chapitre. En parallèle, la genèse de nouvelles mitochondries diminue corrélativement à l’affaiblissement de l’activité de l’AMPK[8] [9].
L’altération du proteome du cytosol et le déclin des enzymes du lysosome empêchent la dégradation complète des protéines et des lipides oxydés et dysfonctionnels. La diminution de la production d’ATP, l’augmentation de la production des espèces réactives de l’oxygène, la formation accrue de lipofuscine aboutissent à l’affaiblissement du lysosome et finalement à l’apoptose, le suicide cellulaire.
Ces dysfonctionnements cellulaires conduisent inévitablement la fonction vers son déclin, particulièrement celle des cellules à durée de vie longue, comme les neurones, les cellules cardiaques et les fibres musculaires squelettiques. La détérioration de l’adaptabilité de l’organisme âgé, qui se caractérise par sa fragilité, sa difficulté à se réparer, le conduit au décès[10].
Au décours de l’enchaînement des causes et des conséquences dans la dégradation du processus de maintenance cellulaire, il apparaît que le dysfonctionnement des mécanismes de renouvellement mitochondrial -dû au ralentissement de l’autophagie- est le processus même du vieillissement[11].
Ce mécanisme retentit à son tour sur le système immunitaire et la réaction inflammatoire. C’est l’objet du prochain chapitre…
Pour en savoir plus, notre ouvrage sur l’avenir du vieillissement: “Vieillir, un destin remédiable“.
[1] Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal. 2010;12(4):503-535. doi:10.1089/ars.2009.2598
[2] Khrapko K. Bodyak N. Thilly WG. van Orsouw NJ. Zhang X. Coller HA. Perls TT. Upton M. Vijg J. Wei JY. Cell-by-cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions. Nucleic Acids Res. 1999;27:2434–2441.
[3] Eskelinen EL. Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009;1793:664–673.
[4] Cuervo AM. Autophagy and aging: keeping that old broom working. Trends Genet. 2008;24:604–612.
[5] Jolly RD. Douglas BV. Davey PM. Roiri JE. Lipofuscin in bovine muscle and brain: a model for studying age pigment. Gerontology. 1995;41:283–295.
[6] Cuervo AM. Autophagy and aging: keeping that old broom working. Trends Genet. 2008;24:604–612.
[7] Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. Mitochondrial Turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Antioxidants and Redox Signaling. 2010;12(4):503–535.
[8] Reznick RM. Zong H. Li J. Morino K. Moore IK. Yu HJ. Liu ZX. Dong J. Mustard KJ. Hawley SA. Befroy D. Pypaert M. Hardie DG. Young LH. Shulman GI. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5:151–156.
[9] Rohrbach S. Niemann B. Abushouk AM. Holtz J. Caloric restriction and mitochondrial function in the ageing myocardium. Exp Gerontol. 2006;41:525–531.
[10] Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal. 2010;12(4):503-535. doi:10.1089/ars.2009.2598
[11] Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal. 2010;12(4):503-535. doi:10.1089/ars.2009.2598