
Qu’est-ce que le vieillissement?
On peut définir le vieillissement comme l’augmentation de la probabilité de mourir ou de faire défaut avec le temps[1], ce qui assimile le corps à une machine qui “s’use” avec le temps. Le vieillissement est considéré par la théorie dominante comme le déclin progressif des fonctions, sous l’effet accumulé de dommages cellulaires et moléculaires causés par les radicaux libres, les agents pathogènes, les toxines, les agents carcinogènes, les erreurs de réplication et de traduction de l’ADN, les dysfonctions protéiques et les forces mécaniques. Cette théorie considère que le coût de la maintenance de l’organisme devient progressivement insoutenable, et que l’organisme est peu à peu submergé par ses défaillances.
Quel est le processus du vieillissement?
Une des interrogations décisives sur le vieillissement concerne la nature des mécanismes qui le provoquent. Est-ce un programme codé dans le génôme qui décide de notre longévité, ou bien s’agit-il des conséquences des aléas de nature physico-chimiques qui endommagent le fonctionnement cellulaire?
Une théorie du vieillissement doit pouvoir également rendre compte de la variabilité de la longévité entre les espèces. Selon Barja[2], il y aurait une multiplicité d’effecteurs du vieillissement car aucun ne peut rendre compte à lui seul de la variabilité inter-espèces en longévité. A partir de là il serait vain d’attendre un gain de longévité à partir d’un effecteur isolé, comme un antioxydant[3].
A priori, selon Hayflick, le vieillissement ne peut provenir que d’un programme défini par des gènes, ou par hasard, du fait d’événements accidentels. Le vieillissement serait soit le prolongement du programme génétique qui dirige tous les processus biologiques depuis la naissance jusqu’à la maturation reproductive, soit le résultat d’une accumulation de pertes aléatoires et irréparables.

Selon Hayflick, il faut d’abord différencier 4 aspects communément confondus: le vieillissement, la détermination de la longévité, les pathologies associées à l’âge, et la mort. En fait, la formulation de la question du vieillissement à partir de ces données préfigure les réponses possibles. Suivant que l’on distingue ou pas vieillissement et longévité, vieillissement et pathologies liées à l’âge, on présuppose différents processus à l’origine des phénomènes.
Barja et d’autres auteurs soutiennent la thèse d’un programme génétiquement déterminé qui fixerait la longévité. Il s’appuie sur des travaux réalisés sur le matériel génétique. La première mutation génétique qui a augmenté la longévité de la souris a été découvert en 1996[4]. La suppression des gènes impliqués dans la longévité a généralement pour effet de la prolonger, ce qui implique que leur action vise à la restreindre[5]. Ces gènes ont été conservés par l’évolution depuis les levures jusqu’aux mammifères et à l’homme[6], ce qui en fait un trait ancien de la cellule eucaryote[7]. La moitié de ces gènes semblent n’avoir aucune autre fonction que d’induire le vieillissement. Ils proviennent pour la plupart du noyau. Le mieux connu est celui qui code la voie de signalisation “GH-insuline/IGF1-like”. Il est déjà présent chez des unicellulaires comme les levures, et sa fonction primordiale pourrait être de contrôler le vieillissement plutôt que le taux de glucose. Cette dernière fonction n’est devenue nécessaire que 500 million d’années plus tard, quand les multicellulaires ont eu besoin de réguler leur taux de glucose et que l’insuline est apparue. La majorité des gènes impliqués dans la longévité dirigent des actions pro-âge, tandis que le seul mécanisme biologique anti-âge reconnu correspond à l’autophagie.
Hayflick défend la thèse du hasard, ou plutôt d’un déterminisme physico chimique décorrélé de toute programmation. Si l’on parle ici de programme plutôt que de but, ou d’intention, c’est que rien ne permet de supposer que la nature suive des plans tracés en vue d’un objectif prédéfini. La théorie darwinienne soumet plutôt les variations biologiques produites par les mutations aléatoires à la pression de la sélection naturelle, qui s’exprime par la capacité d’un organisme à se reproduire, en fait à survivre assez longtemps pour se reproduire. Ainsi, un programme de vieillissement ne signifie pas que la nature a “décidé” qu’il fallait viellir, mais plutôt qu’un organisme qui en serait doté disposerait d’un avantage adaptatif – ce qui au demeurant paraît assez douteux.
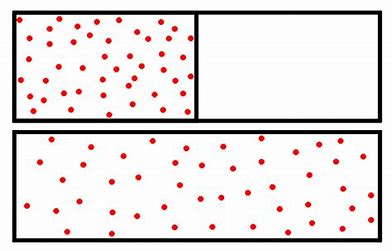
la répartition d’un gaz d’un volume restreint à un volume étendu
C’est bien le hasard, sous la forme de l’entropie, selon Hayflick, qui semble être à l’origine du vieillissement, et qui produit par accumulation de défaillances, une submersion progressive du système de maintenance cellulaire[8]. Le monde non vivant est lui aussi soumis à des changements permanents, non programmés. Le monde vivant comme le non vivant sont soumis à l’entropie, à la seconde loi de la thermodynamique, qui prévaut dans un système ouvert comme dans un système fermé.
Le vieillissement concerne tous les multicellulaires qui atteignent une taille fixe à l’âge de maturité reproductive. Il concerne toutes les espèces, et tous les membres d’une espèce qui ont dépassé l’âge de maturité reproductive; il concerne même les espèces domestiques protégées de la vie sauvage, y compris lorsqu’elles n’ont jamais fait l’expérience du vieillissement sur des millions d’années; il apparaît dans la matière animée comme inanimée, et provient selon Hayflick d’une cause universelle, qui est l’instabilité thermodynamique.
Par ailleurs il est peu probable que le vieillissement soit programmé, au regard de la sélection naturelle, au regard de la diversité des espérances de vie d’individus d’une même espèce, dans un environnement comparable, ce qui est peu compatible avec un programme déterministe[9]. La sélection naturelle n’agit qu’au niveau du succès reproductif d’un individu. Un faible nombre d’individus atteint l’âge de vieillesse dans la nature, et la pression sélective a peu de chance d’influer sur les mutations aléatoires d’un programme de vieillissement.[10] Les mutations qui s’expriment tardivement dans la vie ne sont pas sujettes à sélection[11]. Or la plupart des mutations sont défavorables à l’organisme, et tendent à déstabiliser l’organisme et faciliter son vieillissement. L’hypothèse de pléiotropie antagoniste de Williams postule que certains allèles qui produisent un phénotype avantageux pour la reproduction deviennent inadaptés avec l’âge, et échappent à la pression sélective car ils s’expriment après la période reproductive[12].
Le vieillissement serait alors plutôt lié aux défaillances progressives des mécanismes de protection, plutôt qu’à une auto-destruction anticipée, ces défaillances entraînant une perte de la fonction cellulaire, puis de l’équilibre homéostatique de l’organisme et enfin la mort.
La théorie entropique considère l’organisme comme une machine chimique, qui dépense beaucoup d’énergie pour maintenir son état loin de l’équilibre. C’est par la force des liens biochimiques, que la cellule fait obstacle à l’entropie, au moins jusqu’à l’âge de la maturité reproductive. Des mécanismes de réparation et de remplacement maintiennent l’équilibre du fonctionnement cellulaire. Par la suite, la baisse du niveau d’énergie des molécules, sous l’effet de la dispersion entropique, produit des dysfonctionnements voire une inactivation des biomolécules. Celles-ci ne sont plus réparées ni remplacées, car le dispositif de réparation et de remplacement subit les mêmes altérations que les biomolécules dont il a la maintenance. Il n’y a pas d’immortalité cellulaire, et la seule propriété biologique relativement durable est le message codé dans les molécules contenant de l’information, qui sont soumises à mutations. Selon Kirkwood, le principe de parcimonie explique que le coût métabolique d’une meilleure lutte contre le vieillissement s’effectuerait au détriment de l’efficacité[13]physiologique. On retrouve ce mécanisme dans la gestion des erreurs de réplication de l’ADN et de la synthèse protéique: il existe un optimum sélectif qui minimise le coût énergétique tout en maintenant le taux d’erreur à un niveau acceptable[14].

Au sein d’un organisme multicellulaire, la répartition des fonctions entre soma et lignée germinale permet de maintenir le coût de sélection à un niveau économiquement admissible. L’investissement de ressources abondantes dans la lignée germinale est suffisant pour éviter une détérioration génération après génération, qui entraînerait l’extinction de l’espèce. En revanche, les cellules somatiques sont moins protégées, et la théorie somatique postule que les tissus somatiques sont maintenus assez longtemps pour leur permettre de survivre pendant la période reproductive.
On distingue plusieurs cas de mutations, auxquelles sont exposées les cellules au cours du temps. Celles qui affectent la lignée germinale sont héritables et d’action soit tardive, soit précoce, mais celles qui agissent précocément sont soumises à la pression de sélection. Celles qui affectent la lignée somatique ne sont pas transmissibles d’une génération à l’autre, et celles qui affectent l’ADN mitochondrial non plus. Ces mutations peuvent faciliter la formation de métabolites déstabilisants, commes les ERO, ou entrainer la perte de fonction protectrice de certaines enzymes, ou de leur régulation. Mais seules les mutations germinales précoces sont affectées par la pression sélective et susceptibles d’être renforcées au cours de l’évolution.
La théorie dominante prone que le vieillissement est causé par une accumulation aléatoire de dommages, liés à un défaut du système de maintenance, qui devient trop coûteux[15]. En pratique, la théorie entropique s’appuie sur la théorie de l’oxydation[16] proposée par Harman. Les espèces réactives de l’oxygène (ERO) sont les produits du métabolisme et altèrent les biomolécules. Ce sont les seules molécules capables de briser la liaison covalente au sein des biomolécules. Produites en excès, elles peuvent raccourcir la durée de vie de l’organisme. Pour autant, aucune étude n’a montré une augmentation de la durée de vie à ce jour grâce aux antioxydants[17]. La théorie du stress oxydatif est remise en cause actuellement car la longévité n’est pas corrélée au statut antioxydant[18].
La théorie de l’accumulation des dommages causés sur l’ADN a été dominant dans le champ de la gérontologie. Il y a bien une accumulation des lésions moléculaires avec le temps[19]. La signalisation des altérations de l’ADN peut même être activée sans qu’il y ait réellement un défaut sur l’ADN[20]. Cependant, il n’y a pas de relation linéaire entre le métabolisme mitochondrial et le dommage oxydatif[21]. Cette théorie est très controversée.
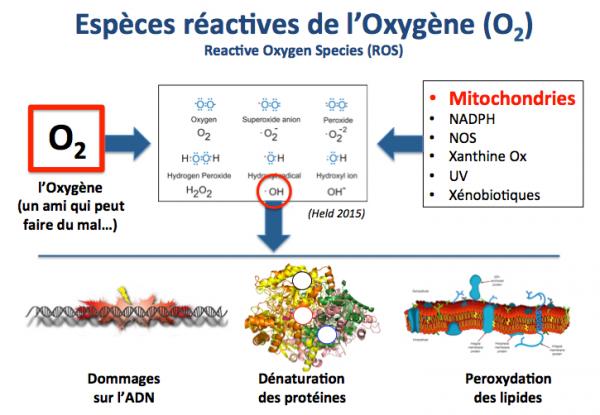
La théorie de l’accumulation des dommages sous l’effet des espèces réactives de l’oxygène (ERO) a permis aux tenants de la thèse du hasard de remporter une bataille théorique contre la théorie de la programmation du vieillissement. Mais la théorie des radicaux libres qui vient appuyer la thèse de l’entropie n’est pas confirmée[22]. Certes, des niveaux modérés d’ERO contribuent à l’adaptation (hormésis) de la régulation d’enzymes protectrices qui participent au dispositif de longévité. Pourtant, la théorie de l’oxydation a fléchi à son tour devant certains faits: d’une part, aucun antioxydant n’a pu augmenter la durée de vie d’organismes de n’importe quelle espèce. Au contraire, certaines études ont montré que les antioxydants pouvaient être toxiques. D’un autre côté, la longévité est associée à une moindre production d’ERO[23]. Toutefois, des contre-exemples affichent une décorrélation entre la durée de vie et le niveau de dommage oxydatif[24]. Des taupes ont une longévité élevée, bien qu’un statut oxydatif également élevé. Les mutations de l’ADN mitochondrial (mtADN) sont plutôt liées à des erreurs de réplication qu’aux radicaux libres; et les mutations de l’mtADN présentent peu d’effets au regard de leurs nombreuses copies au sein de la cellules[25]. Chez l’homme, l’effet des antioxydants sur les pathologies chroniques s’est avéré imprévisible, et parfois toxique[26]. Enfin, le mécanisme protecteur qui devait rendre compte du mode d’action des antioxydants, l’hormésis, a fait long feu.
L’hormésis est-elle utile contre le vieillissement?
L’hormésis correspond à l’induction d’une enzyme de détoxification en présence d’une substance toxique (endogène ou exogène), qui offre en retour une protection à l’organisme contre une dose supérieure, potentiellement léthale de la substance[27][28][29].
L’hormésis, pour expliquer l’augmentation de la longévité en présence de stress modérés et répétés, a été invoquée comme une stimulation de la réponse du système de maintenance et de réparation cellulaire, et comme une cause d’adaptation cellulaire caractérisée par la capacité à endurer des stress plus importants. Mais la compréhension des processus biologiques requiert la connaissance des réactions biochimiques sous-jacentes, en plus des informations sur les circuits de régulation génétique qui affectent ces réactions.
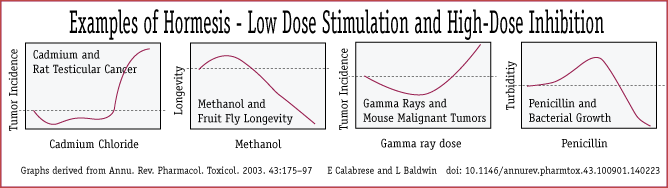
La première étude qui a montré la possibilité d’allonger l’espérance de vie date de 1988[30]. On observe que des stress modérés, comme la restriction calorique, le stress oxydatif, augmentent l’espérance de vie dans différentes espèces [31]. La restriction calorique cible une réponse adaptative ancienne de l’évolution aux changements de l’environnement, permettant une allocation des ressources vers la maintenance somatique plutôt que vers l’anabolisme ou la reproduction.
L’exposition répétée à des chocs thermiques augmente la durée de vie chez la drosophile[32]. La restriction calorique a été décrite comme un stresseur modéré. Les ERO seraient également des stresseurs qui stimuleraient les mécanismes de résistance aux ERO[33]. Cependant, cette adaptation supposée en vue d’un stress majeur ne répond pas au problème posé par le vieillissement. Celui-ci n’est pas le fruit d’un stress majeur, mais d’un stress mineur répété quotidiennement sur une longue durée. L’hormésis serait alors inutile, car elle n’offrirait pas de protection vis à vis des stresseurs de bas niveau qui induisent le vieillissement.
Si l’hormésis induit des dommages et ralentit le vieillissement, alors celui-ci ne dépend pas de ces dommages[34].Ce qui invalide la théorie de l’accumulation des dommages moléculaires[35]. La théorie du vieillissement par oxydation[36] est finalement invalidée avec son argument principal, l’hormésis. Il apparaît que la restriction calorique ne réduit pas les mutations spontanées mais néanmoins augmente la durée de vie, et agit donc sur la longévité plutôt que sur le vieillissement[37].
En conclusion, ce ne sont pas les dommages qui font vieillir les organismes, ils ne vivent pas assez longtemps pour cela[38]. Si les humains meurent de pathologies liées à l’âge, causées par des processus cellulaires actifs, sommes nous pour autant en face d’un programme, comme le suppose Barja, qui fixerait la longévité, indépendamment des aléas du vieillissement entropique?
La longévité est-elle prédéfinie?
“L’entropie explique le vieillissement, le déterminisme génétique explique la longévité, et une terminologie approximative explique l’incompréhension des deux” disait Hayflick[39]. Autrement dit, il y aurait d’un côté des dommages de nature essentiellement chimique et physique, et d’un point de vue biologique peu ou pas régulés, qui entraîneraient une perte progressive de fonction, et une capacité diminuée à supporter les contraintes endogènes et environnementales. D’un autre côté se trouveraient des processus régulés censés prévenir, réparer, ou limiter le dommage. L’équilibre entre ces deux forces détermine l’espérance de vie de l’individu et la trajectoire de mortalité d’une population.
La longévité, à la différence du vieillissement est gouvernée par le génome. Elle correspond à l’état de fonctionnement optimal des molécules, lié à l’efficacité des dispositifs de maintenance, gouvernés par le génome. Celui-ci dirige le destin biologique jusqu’à la maturité reproductive, selon les modalités sélectionnées sous la pression de la sélection naturelle. Indirectement, le génome prévoit donc la longévité.
Pour reprendre la formule imagée de Hayflick, disons que la détermination de la longévité est un processus anabolique gouverné par le génome, qui correspond à une tendance à durer dans le temps, tandis que le vieillissement est un processus aléatoire de type catabolique qui correspond à une tendance au changement.

La différence d’espérance de vie entre espèces, après correction des masses corporelles et des sources externes de mortalité, indique que la durée de vie est sous contrôle génétique, et sujette à évolution[40]. Ainsi, soit il y a un programme génétique qui définit le format de durée de vie de l’espèce, soit le vieillissement correspond au débordement des dispositifs de protection contre le vieillissement génétiquement programmés.
Des facteurs génétiques, épigénétiques et cellulaires interviennent dans la longévité. La signalisation depuis les gonades contrôle la longévité via de multiples familles de facteurs de transcription, et implique la régulation du métabolisme du glucose et des lipides. Les gènes jouent un rôle majeur dans la détermination de la longévité, pour autant déterminent-ils les changements liés à l’âge?
Barja avance plusieurs arguments en faveur de la programmation de la longévité par une action directe sur le vieillissement[41]:
Il pose que le programme de vieillissement prolonge le programme de développement et de maturation de l’organisme, et observe que les animaux à longévité prolongée bénéficient d’une moindre activité des facteurs de vieillissement plutôt que d’une meilleure défense anti-âge.
Ces facteurs de vieillissement sont exprimés sous la dépendance des gènes qui les programment et correspondent à différents mécanismes: la production de radicaux libres par la mitochondrie, la peroxidation lipidique membranaire, l’apoptose, la réduction de la longueur des télomères, l’inclusion de fragments de mtADN dans le nADN, l’inflammation et les facteurs épigénétiques. Des acides aminés comme la méthionine et le tryptophane[42] semblent intervenir sur l’extension de la longévité. Il n’existe qu’un mécanisme anti-âge caractéristique des espèces à longévité élevée, il s’agit de l’autophagie.
Barja distingue trois principaux mécanismes effecteurs du vieillissement, sur lesquels l’environnement peut influer et qui sont susceptibles d’augmenter la longévité: la production des radicaux libres mitochondriaux peut être diminuée, la concentration en acides gras polyinsaturés dans la membrane cellulaire peut être réduite en faveur d’acide gras saturés, et l’autophagie, système de nettoyage des déchets cellulaires, peut être stimulée. Les radicaux libres mitochondriaux peuvent augmenter l’insertion de fragments d’mtADN dans l’ADN nucléaire (nADN), ce qui produit des aneuploidies pendant la mitose, corrompt les gènes, arrête la division cellulaire et promeut une instabilité génomique[43]. L’instabilité de la membrane cellulaire riche en acides gras insaturés la rend perméable aux ERO, qui peuvent dès lors atteindre le noyau. La présence de radicaux libres mitochondriaux entraine une peroxydation lipidique des membranes cellulaires, notamment les membranes mitochondriales. Cette réaction en chaine génère des produits toxiques et mutagènes, qui peuvent traverser le noyau par diffusion et réagir avec les groupes aminés de l’ADN et des protéines, entraînant des dommages dans l’ADN nucléaire et mitochondrial[44]. La diminution de l’autophagie conduit à l’accumulation de lipides et de protéines peroxydés, à la diminution de l’élimination des mitochondries altéres par les ERO, et à l’accumulation de déchets cellulaires, sous forme de lipofuscine.
Il s’agit alors pour des facteurs anti-âge de produire moins de radicaux libres au niveau mitochondrial, et d’obtenir des membranes cellulaires plus résistantes aux ERO. Ces processus diminuent dans quatre cas de manipulation nutritionnelle et pharmacologique qui augmentent la longévité des mammifères: la restriction calorique, la restriction en methionine, la restriction protéique, et la rapamycine[45].
Ces facteurs anti-âge, la rapamycine, la restriction calorique et la suppression de la méthionine s’effectuent en partie en agissant sur les mécanismes exécutifs du vieillissement, étiquetés comme pro-âge, mais majoritairement en modifiant l’expression de gènes nucléaires[46] impliqués dans la longévité, ce qui modifie l’activité des effecteurs en aval et renforce l’action protectrice des facteurs anti-âge. Cette action dominante sur les gènes vient en appui d’une théorie programmatique de la longévité.
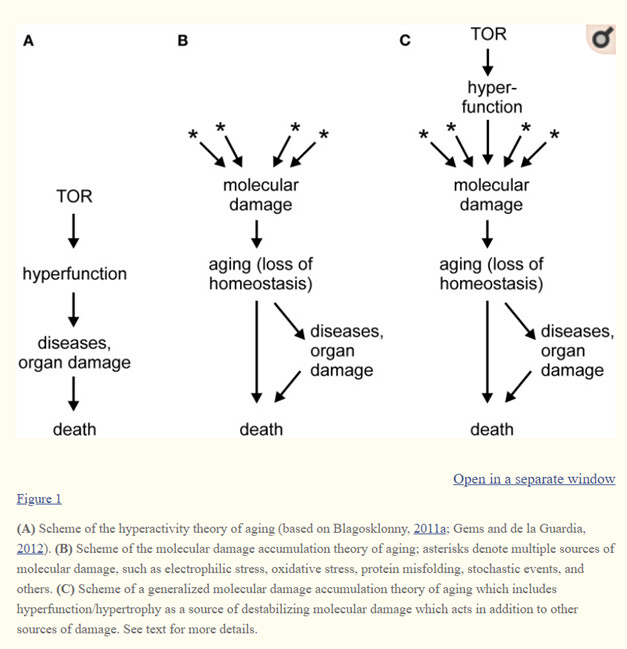
Il y a bien une corrélation inverse entre le taux de radicaux libres mitochondriaux, le niveau d’insaturation de la membrane cellulaire et la longévité des espèces. Mais pour l’instant rien n’affirme qu’il existe un lien entre l’autophagie, l’apoptose, la protéostasie[47], la longueur des télomères[48], l’inflammation, la concentration de fragments de mtADN dans l’nADN, ou des marqueurs épigénétiques et la longévité.
Par ailleurs, les modifications dans l’expression des gènes sous l’effet de la restriction calorique sont spécifiques aux espèces et aux tissus. La longévité se modifie sous l’effet de facteurs qui modifient l’expression des gènes, ce qui expliquerait la variabilité entre les individus d’une même espèce, et entre les espèces. Les gènes de l’âge peuvent également être modifiés par des facteurs épigénétiques, comme la methylation de l’ADN, l’acétylation de l’histone, certains auteurs supposent même l’existence d’une horloge épigénétique.[49]

L’augmentation de la longévité sous l’effet de la restriction calorique traduit bien des changements dans le dispositif génomique de la longévité plutôt que dans les mécanismes du vieillissement[50]. La restriction calorique est la méthode environnementale d’augmentation de la longévité la plus efficace à ce jour[51]. Elle permet également de retarder l’apparition de maladies dégénératives chez les mammifères[52]. Chez l’homme, la restriction calorique améliore également les facteurs métaboliques et hormonaux impliqués dans la pathogenèse du diabète de type 2, des maladies cardio-vasculaires et du cancer, réduit le stress oxydatif s[53]. La restriction calorique et en methionine réduisent le taux de fuite de radicaux libres, en sept semaines et aussi longtemps que dure la cure[54]. La fuite de radicaux libres dans la mitochondrie à l’occasion de la production d’énergie dans le cycle de Krebs est donc un phénomène régulé, par voie génétique, ou épigénétique -grâce à la restriction calorique et l’exercice aérobie-, et non d’un effet collatéral contingent de la chaîne respiratoire mitochondriale.
Il existe une détermination génétique du taux de fuite de radicaux libre dans les mitochondries, qui affecte la longévité des espèces, indépendamment de leur taille et de leur masse. Celle-ci explique la longévité exceptionnelle de certains pigeons (30 ans) par rapport aux rats de même poids (quatre ans)[55]. Le taux de fuite de radicaux libres peut être amélioré par l’exercice aérobie sur les tissus concernés (muscles et coeur) et augmenter l’espérance de vie moyenne des individus qui le pratiquent[56] . Cet effet est contrebalancé par la production cytosolique de radicaux libres dont les effets sont systémiques[57]. Le pourcentage d’électrons convertis à la production d’ERO dans la mitochondrie n’est donc pas fixe et dépend à la fois d’un programme génétique, et de facteurs épigénétiques. Au final, les signaux afférents, les gènes pro-âge et les effecteurs du vieillissement collaborent au cadrage d’une longévité pour une espèce donnée[58].
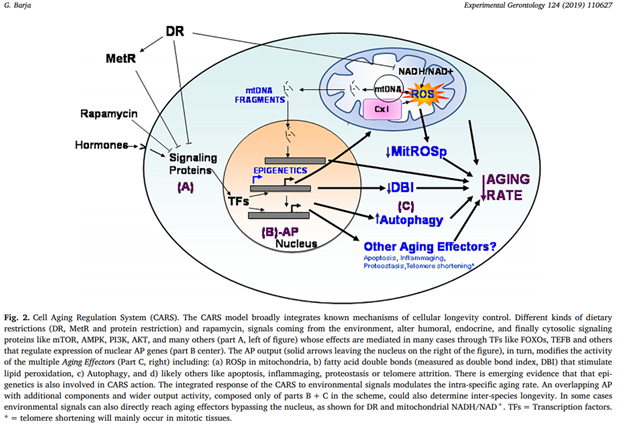
La longévité d’une espèce dépend in fine de l’activité des mécanismes qui produisent le vieillissement, qui sont définis génétiquement pour chaque espèce. Ces mécanismes sont sensibles à des influences environnementales, qui modulent les mécanismes intrinsèques du vieillissement et la durée de vie des individus. Les mécanismes en question concernent la mitose et l’inclusion de fragments d’ADN mitochondrial dans l’ADN nucléaire. La réduction de la longueur des télomères, l’apoptose et l’arrêt de division cellulaire sont des mécanisme de vieillissement accessoires de la mitose. Ils ne concernent que les cellules qui se divisent fréquemment, comme les cellules de la peau et du colon. En revanche, ces mécanismes ne concernent que marginalement le cerveau, le muscle squelettique, le coeur, qui se divisent peu ou pas. Ces cellules sont exposées aux radicaux libres produits par les mitochondries, à la peroxidation lipidique liée au taux d’acides gras insaturés dans la membrane cellulaire, et à l’autophagie, comme dans les cellules mitotiques. Certains auteurs posent que ces mécanismes relèvent d’un programme génétique, et que la longévité humaine est fixée à 122 ans. Des premiers primates aux hommes, la longévité a été multipliée par 10[59].
Le vieillissement ne nécessite pas de gènes pour s’exprimer. L’entropie suffit. Chez les unicellulaires, le vieillissement a été défini comme le nombre de cellules filles produites à partir d’une cellule mère avant sénescence[60]. Chez les multicellulaires ce sont les déterminants de la longévité qui sont étudiés plutôt que ceux du vieillissement. Celle-ci paraît gouvernée par un programme génétique qui définit les processus de développement et de maturation des organismes. Qu’advient-il de ce programme de croissance qui gouverne la maturation des organismes jusqu’à l’âge de maturité reproductive, après que celui-ci a été atteint? Les pathologies qui apparaissent avec l’âge sont-elles secondaires à sa disparition, ou à son expression prolongée? Comment s’articulent les pathologies liées à l’âge et le programme de longévité?
Les maladies associées au vieillissement
Le troisième aspect de la finitude du vivant selon Hayflick correspond aux pathologies associées à l’âge. Celles-ci n’ont pas les propriétés du vieillissement décrites plus haut. Hayflick rend compte des pathologies liées à l’âge par voie de conséquence des mécanismes du vieillissement, secondaires à l’entropie, à l’accumulation des défaillances métaboliques. La vulnérabilité et les pathologies associées à l’âge apparaissent quand la perte de fidélité moléculaire excède la capacité de réparation[61]. Selon la théorie de l’entropie, le vieillissement n’est pas une maladie mais accroît la vulnérabilité aux maladies. Celles-ci ne permettent alors pas de comprendre le mécanisme du vieillissement en tant que tel. Ainsi aucun progrès en médecine gériatrique n’apporterait de connaissance sur les mécanismes fondamentaux du vieillissement[62] selon Hayflick.
Pour autant, le vieillissement[63]de l’organisme comme affaiblissement de l’organisme et l’apparition des pathologies sont intimement corrélés. L’immunité innée d’abord, puis l’acquise se détériorent pendant le vieillissement par perte de la capacité de reconnaissance des agents pathogènes. La perte de la protéostasie, produisant une accumulation de protéines et d’organites dans la cellule est caractéristique du vieillissement. Les voies de réponse au stress, les mécanismes d’autophagie, et le système ubiquitine-protéasome sont dégradés[64]. L’altération du génome est également impliquée dans le vieillissement. Des agressions d’origine physique, chimique et biologique, ainsi que des mécanismes endogènes y participent. La diminution de la longueur des télomères à l’extrêmité des chromosomes contribue à l’arrêt de croissance des cellules sénescentes. Les souris avec de plus long télomères ont une durée de vie supérieure à celles aux télomères plus courts. Le vieillissement se manifeste in fine par des pathologies, cancers, infarctus du myocarde, accidents vasculaires, cécité, qui traduisent sous forme critique, et létale, le dysfonctionnement sous-jacent. Comment rendre compte de la pathogenèse de l’athérosclérose, du diabète, des cancers, des maladies neurodégénératives, sans impliquer les mécanismes sous-jacents qui altèrent l’équilibre de l’organisme jusqu’à l’expression pathologique et la mort?
Le programme de croissance: la voie mTOR
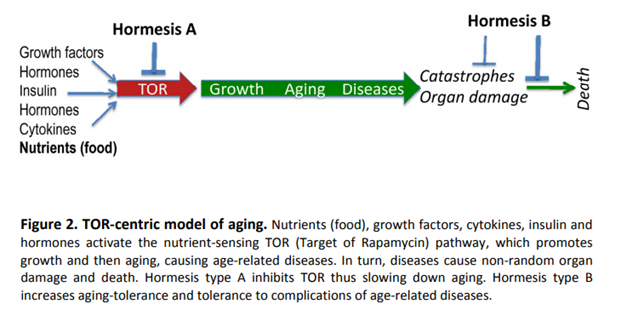
La caractéristique principale du vieillissement cellulaire, quand on observe les cellules des différents tissus altérés par l’âge, est l’hypertrophie, l’hyperfonction, comme le phénotype pro-inflammatoire, et la résistance au signal régulateur, comme la résistance à l’insuline. Selon Blagosklonny, c’est cette transformation qui entraine une rupture de l’homéostasie, les maladies et la mort[65]. L’hyperfonction est spécifique à chaque tissu: la résorption osseuse est le fait des ostéoclastes, le tonus artériel est produit par les cellules musculaires lisses, qui conduisent à l’ostéoporose et à l’hypertension.

A partir de ces observations, des chercheurs ont mis en cause le programme de croissance de l’organisme, dont la persistance au-delà de la maturité sexuelle pourrait devenir toxique pour l’organisme exposé aux effets de l’entropie. La théorie de l’hyperfonction n’est ni une théorie du vieillissement programmé, ni une théorie du vieiillissement aléatoire. Elle pose que les mécanismes du vieillissement sont ceux de la croissance, de la différentiation et du développement. Elle part de l’observation de cultures cellulaires. Le vieillissement n’est pas produit par les dommages moléculaires liés au hasard, mais par l’hyperfonction et l’hypertrophie cellulaire secondaires à une persistance inappropriée à l’âge adulte des programmes de développement, notamment la voie de signalisation mTOR. Les cellules finiraient bien par être mises en défaut par l’entropie sous l’effet d’un temps suffisamment long, mais l’action nocive de la voie mTOR devance les effets toxiques des ERO[66].
Les gènes de la croissance[67] encodent les voies de signalisation type insuline/PI3K/TOR[68]. Ces voies sont essentielles à la croissance et à la survie pendant la période de croissance, puis au vieillissement et aux maladies; elles participent à la puberté et à la ménopause. Le programme génétique de la croissance vise l’accomplissement du développement et la reproduction. Mais la nature n’a pas intégré la mise en sommeil de ce dispositif après l’atteinte des objectifs. Le vieillissement est le prix de la croissance.
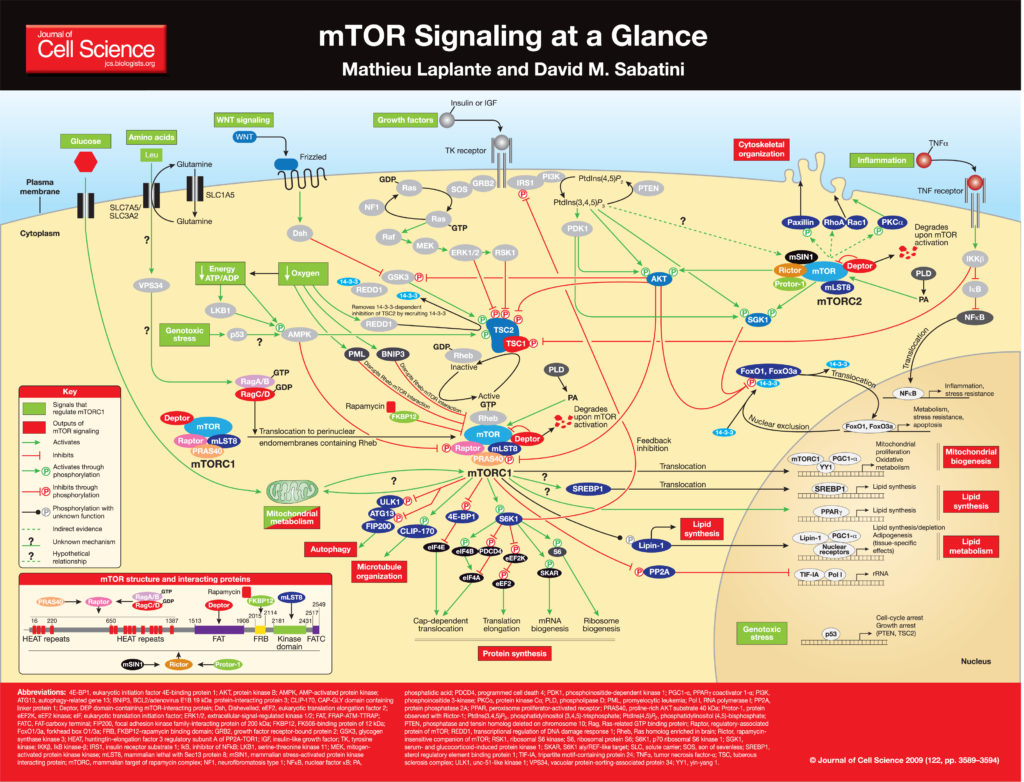
La voie de signalisation nutritionnelle mTOR est activée par l’insuline, les facteurs de croissance et les aliments. Elle accroît la synthèse protéique, stimule la biogenèse des ribosomes, et la croissance cellulaire, inhibe l’autophagie, induit l’accumulation de protéines agrégées, accroît la sécrétion de facteurs de croissance, et conduit à la résistance à l’insuline et au facteur de croissance[69].
mTOR intègre les signaux émis par l’insuline, les cytokines, les mitogènes, l’O2 et les nutriments. Le glucose active la voie mTOR, qui accroît la production d’insuline, qui stimule en retour la production de mTOR, produisant une résistance à l’insuline et à l’IGF1. Cette résistance accroît le niveau de glucose, ce qui conduit au diabète de type 2, à l’insuffisance rénale et à la mort.
Les facteurs de croissance stimulent à la fois la croissance cellulaire et sa division, activent la progression du cycle cellulaire. Lorsque le cyle cellulaire est bloqué, les facteurs de croissance qui stimulent toujours la voie mTOR stimulent la croissance cellulaire, alors que celle-ci ne peut plus se diviser. Dans le même temps, mTOR/S6K bloquent les récepteurs à insuline, et rendent les cellules résistantes à l’insuline. La cellule devient hypertrophique et hyperfonctionnelle, sénescente.
Ainsi, un ADN endommagé ou une radiation peut arrêter le cycle cellulaire sans inhiber la voie signalétique de croissance[70], et l’active plutôt. La présence de mTOR est nécessaire pour la conversion cellulaire sénescente.
La voie mTOR guide la croissance cellulaire. Celle-ci est normalement contrebalancée par la division cellulaire. Chez les cellules au repos, elle conduit à la sénescence. Quand la division est bloquée elle produit un phénotype sénescent, hypertrophique, hyperactif, hyper-fonctionnel, hyper sécrétoire, auquel répond une résistance compensatrice des récepteurs (insuline), et une activation lysosomale compensatrice[71].
sous l’effet des ostéoclastes
Lorsque la croissance est terminée, la géroconversion correspond au prolongement de celle-ci. mTOR provoque l’expression du phénotype sécrétoire sénescent, ce qui conduit aux pathologies liées à l’âge. Le vieillissement est un “quasi programme” qui prolonge le programme de croissance cellulaire, dirigé notamment par mTOR[72]. La sénescence cellulaire est définie comme un arrêt du cycle cellulaire irréversible, causé par une altération de l’ADN ou un autre stress. Mais le vieillissement n’est pas causé par l’épuisement de la prolifération cellulaire et l’arrêt du cycle cellulaire. C’est l’évitement de l’arrêt du cycle qui conduisent à l’hyperplasie cellulaire et au cancer[73].
Le vieillissement est compris comme une forme de croissance découplée du cycle de division cellulaire. Lorsque la voie de signalisation mTOR ne peut plus conduire une croissance normale, elle conduit au vieillissement. Ce mécanisme est commun aux levures, aux vers comme aux mammifères.
La connaissance des mécanismes de croissance permet d’interpoler ceux du vieillissement. La protéine P53 et l’hypoxie inhibent mTOR, ce qui supprime la géroconversion des cellules. On peut établir une cartographie des réseaux suivant leur influence sur le vieillissement. Les gènes qui activent la voie mTOR sont des gérogènes comme le récepteur à l’insuline, PI-3K, Akt), tandis que ceux qui l’inhibent sont des gérosuppresseurs (AMPK, PTEN, TSC)[74].
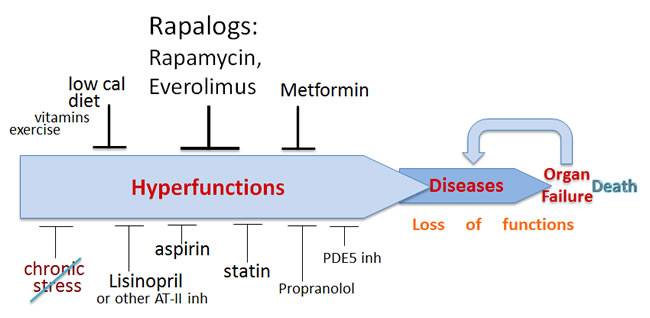
Les aliments, les facteurs de croissance, les hormones et les cytokines activent une voie de signalisation cellulaire stimulant la croissance, appelée mTOR pour (Target Of Rapamycin). mTOR stimule la croissance et l’anabolisme celllulaire, inhibe l’autophagie et accroît les fonctions cellulaires. Les cellules croissent en taille, évoluent dans leur cycle cellulaire et se divisent. En l’absence de ces facteurs de croissance, les cellules deviennent quiescentes. Mais sous l’influence des facteurs de croissance, lorsque leur cycle est bloqué, la voie de mTOR cause la sénescence cellulaire[75]. Celle-ci serait donc une continuation de la différentiation cellulaire[76]. Certaines fonctions sont alors amplifiiées, comme la production d’hormones, d’enzymes, de métabolites, suivant le type cellulaire, ou l’agrégation plaquettaire, ou la résorption osseuse par les osteoclastes, ou l’hypersécrétion des fibroblastes, ou la contraction des muscles de la paroi artérielle, la lipogenèse par les adipocytes, l’inflammation par les neutrophiles, la phagocytose par les macrophages, ce qui entraîne l’hypertrophie des organes et leur fibrose, l’athérosclérose et l’hypertension. Il n’y a pas de pathologie liée à l’âge qui ne soit causée par l’hyperfonction, médiée par mTOR. La perte de fonction est la phase terminale d’un processus qui s’engage avec l’hyperfonction sénescente[77].

En effet la voie mTOR est impliquée dans de nombreuses pathologies du vieillissement: hypertension, hypertrophie cardiaque, ostéoporose, obésité, ostéoarthrite, perte d’audition, tumeurs bénignes et malignes, cancers, diabète de type 2 et ses complications (rétinopathie et hypertrophie rénale), dégénérescence maculaire liée à l‘âge, athérosclérose, hypertrophie cardiaque, fibrose organique, maladie d’Alzheimer, maladie de Parkinson[78]. mTOR limite la longévité en accélérant les pathologies liées à l’âge. Ces maladies sont les manifestations du vieillissement qui limitent la longévité.
En conclusion
En l’état actuel des connaissances dans le champ de la gérontologie, les rôles respectifs des effets physico-chimiques de l’entropie sur l’homéostasie cellulaire, et du génôme sur la programmation des fonctions cellulaires s’articulent sur la théorie évolutionniste darwinienne. Les organismes exposés aux mutations aléatoires sont soumis à une pression sélective qui retient les variants les plus efficaces pour la survie jusqu’à l’âge de la reproduction. Les programmes génétiques de développement et de maturation sont soumis à cette pression seulement sous ce critère. Ainsi, aucune sélection n’a opéré au cours de l’évolution sur les organismes dont les mutations favorisaient un ralentissement du programme de croissance cellulaire après la maturité sexuelle. Celui-ci agit in fine en facteur limitant de la longévité, à travers les dysfonctionnements qu’il induit dans l’organisme au-delà de l’âge de la maturité. La cellule, exposée aux aléas qui altèrent l’ADN et déclenchent un arrêt du cycle cellulaire pour obtenir la quiescence, subit une croissance forcée sous l’effet du programme de développement, qui conduit la cellule à un fonctionnement pléthorique, découplé de la régulation homéostatique, qui produit des effets délétères sur le fonctionnement des tissus et des organes, jusqu’à la défaillance critique de l’organisme, et à la mort. Bloquer la voie mTOR de croissance cellulaire apparaît après l’atteinte de la maturité comme un moyen de ralentir la géro-conversion cellulaire et le vieillissement. C’est la visée des sénolytiques.
Pour en savoir plus, lisez notre ouvrage sur l’avenir du vieillissement: “Vieillir, un destin remédiable“.

[1] Entropy explains aging, genetic determinism explains longevity, and undefined terminology explains misunderstanding both. Hayflick L PLoS Genet. 2007 Dec; 3(12):e220.
[2] Barja G. Towards a unified mechanistic theory of aging. Exp Gerontol. 2019 Sep;124:110627. doi: 10.1016/j.exger.2019.05.016. Epub 2019 Jun 5. PMID: 31173843.
[3] Pérez, V.I., Bokov, A., Van Remmen, H., et al., 2009. Is the oxidative stress theory of aging dead? Biochim. Biophys. Acta 1790, 1005–1014.
[4] Brown-Borg, H.M., Borg, K.E., Meliska, C.J., Bartke, A., 1996. Dwarf mice and the ageing process. Nature 384 (6604), 33.
[5] Bishop, N.A., Guarente, L., 2007. Genetic links between diet and lifespan: shared mechanisms from yeast to humans. Nat Rev Genet 8, 835–844. B
[6] . Clark, W.R., 2004. Reflections on an unsolved problem of biology: the evolution of senescence and death. Adv. Gerontol. 14, 7–20
[7] Bishop, N.A., Guarente, L., 2007. Genetic links between diet and lifespan: shared mechanisms from yeast to humans. Nat Rev Genet 8, 835–844.
[8] Finch CE, Kirkwood TBL. Chance, development and aging. New York: Oxford University Press; 2000.
[9] Zimniak P. Detoxification reactions: relevance to aging. Ageing Res Rev. 2008;7(4):281-300. doi:10.1016/j.arr.2008.04.001
[10] Medawar PB. An unsolved problem of biology. London: H. K. Lewis; 1952.
[11] Understanding ageing from an evolutionary perspective. Kirkwood TB J Intern Med. 2008 Feb; 263(2):117-27.
[12] Williams GC. Pleiotropy, natural selection, and the evolution of senescence. Evolution. 1957;11:398–411.
[13] Evolution of ageing. Kirkwood TB Nature. 1977 Nov 24; 270(5635):301-4.
[14] Translational accuracy and the fitness of bacteria. Kurland CG Annu Rev Genet. 1992; 26():29-50.
[15] Understanding the odd science of aging. Kirkwood TB Cell. 2005 Feb 25; 120(4):437-47.
[16] “I thought, thought, thought for four months in vain and suddenly the idea came”–an interview with Denham and Helen Harman. Interview by K. Kitani and G.O. Ivy. Harman D, Harman H Biogerontology. 2003; 4(6):401-12.
[17] Myung SK, Ju W, Cho B, Oh SW, Park SM, Koo BK, Park BJ, and Korean Meta-Analysis Study Group. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta-analysis of randomised controlled trials. BMJ. 2013; 346:f10. 10.1136/bmj.f10
[18] Antioxidant defense and aging in C. elegans: is the oxidative damage theory of aging wrong? Gems D, Doonan R Cell Cycle. 2009 Jun 1; 8(11):1681-7.
[19] Molecular damage in cancer: an argument for mTOR-driven aging. Blagosklonny MV Aging (Albany NY). 2011 Dec; 3(12):1130-41.
[20] Pseudo-DNA damage response in senescent cells. Pospelova TV, Demidenko ZN, Bukreeva EI, Pospelov VA, Gudkov AV, Blagosklonny MV Cell Cycle. 2009 Dec 15; 8(24):4112-8.
[21] Food consumption and individual lifespan of adults of the blowfly, Calliphora stygia: a test of the ‘rate of living’ theory of aging. Hulbert AJ, Usher MJ, Wallman JF Exp Gerontol. 2004 Oct; 39(10):1485-90.
[22] Aging: ROS or TOR. Blagosklonny MV Cell Cycle. 2008 Nov 1; 7(21):3344-54.
[23] Free radicals and aging. Barja G Trends Neurosci. 2004 Oct; 27(10):595-600.
[24] High oxidative damage levels in the longest-living rodent, the naked mole-rat. Andziak B, O’Connor TP, Qi W, DeWaal EM, Pierce A, Chaudhuri AR, Van Remmen H, Buffenstein R Aging Cell. 2006 Dec; 5(6):463-71.
[25] Buffenstein, R., Edrey, Y.H., Yang, T., Mele, J., 2008. The oxidative stress theory of aging: embattled or invincible? Insights from non-traditional model organisms. Age 30, 99–109. B
[26] Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C JAMA. 2007 Feb 28; 297(8):842-57.
[27] Hormesis: the dose-response revolution. Calabrese EJ, Baldwin LA Annu Rev Pharmacol Toxicol. 2003; 43():175-97.
[28] Stress-response hormesis and aging: “that which does not kill us makes us stronger”. Gems D, Partridge L Cell Metab. 2008 Mar; 7(3):200-3.
[29] The ecological stress theory of aging and hormesis: an energetic evolutionary model. Parsons PA Biogerontology. 2007 Jun; 8(3):233-42.
[30] A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Friedman DB, Johnson TE Genetics. 1988 Jan; 118(1):75-86.
[31] Radak Z, Chung HY, Goto S. Exercise and hormesis: oxidative stress‐related adaptation for successful aging. Biogerontology. 2005; 6:71‐75.
[32] Hercus MJ, Loeschcke V, Rattan SI. Lifespan extension of Drosophila melanogaster through hormesis by repeated mild heat stress. Biogerontology. 2003; 4:149‐156.
[33] Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007; 6:280‐293.
[34] Hormesis does not make sense except in the light of TOR‐driven aging Mikhail V. Blagosklonny
[35] Blagosklonny MV. mTOR‐driven aging: speeding car without brakes. Cell Cycle. 2009; 8:4055‐4059.
[36] Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radic Biol Med. 2011; 51:327‐336.
[37] Edman U, Garcia AM, Busuttil RA, Sorensen D, Lundell M, Kapahi P, Vijg J. Lifespan extension by dietary restriction is not linked to protection against somatic DNA damage in Drosophila melanogaster. Aging Cell. 2009; 8:331‐338.
[38] Blagosklonny MV. Hormesis does not make sense except in the light of TOR-driven aging. Aging (Albany NY). 2011 Nov;3(11):1051-62. doi: 10.18632/aging.100411. PMID: 22166724; PMCID: PMC3249451.
[39] Entropy explains aging, genetic determinism explains longevity, and undefined terminology explains misunderstanding both. Hayflick L PLoS Genet. 2007 Dec; 3(12):e220.
[40] Ageing studies on bats: a review. Brunet-Rossinni AK, Austad SN Biogerontology. 2004; 5(4):211-22.
[41] Barja, G. Towards a unified mechanistic theory of aging. Experimental Gerontology, 124, 110627. https://doi.org/10.1016/J.EXGER.2019.05.016
[42]De Marte ML, Enesco HE. Influence of low tryptophan diet on survival and organ growth in mice. Mech Ageing Dev. 1986 Oct;36(2):161-71. doi: 10.1016/0047-6374(86)90017-5. PMID: 3784629.
[43] Macedo, J.C., Vaz, S., Bakker, B., et al., 2018. FoxM1 repression during human aging leads to mitotic decline and aneuploidy-driven full senescence. Nat. Commun. 9, 2834. https://doi.org/10.1038/s41467-018-05258-6.
[44] Chaudhary, A.K., Nokubo, M., Reddy, G.R., et al., 1994. Detection of endogenous malondialdehyde-deoxyguanosine adducts in human liver. Science 265, 1580–1582.
[45] Hulbert, A.J., Pamplona, R., Buffenstein, R., Buttemer, W.A., 2007. Life and death: metabolic rate, membrane composition, and life span of animals. Physiol. Rev. 87, 1175–1213.
[46] Pan, H., Finkel, T., 2017. Key proteins and pathways that regulate lifespan. J. Biol. Chem. 292, 6452–6460. https://doi.org/10.1074/jbc.R116.771915.
[47] Pickering, A.M., Lehr, M., Miller, R.A., 2015. Lifespan of mice and primates correlates with immunoproteasome expression. J. Clin. Invest. 125, 2059–2068. https://doi. org/10.1172/JCI80514.
[48] Vera, E., Bernardes de Jesus, B., Foronda, M., Flores, J.M., Blasco, M.A., 2012. The rate of increase of short telomeres predicts longevity in mammals. Cell Rep. 2, 732–737. https://doi.org/10.1016/j.celrep.2012.08.023.
[49] Jones, M.J., Goodman, S.J., Kobor, M.S., 2015. DNA methylation and healthy human aging. Aging Cell 14, 924–932. https://doi.org/10.1111/acel.12349.
[50] Recent developments in yeast aging. Kaeberlein M, Burtner CR, Kennedy BK PLoS Genet. 2007 May 25; 3(5):e84.
[51] Anderson, R.M., Weindruch, R., 2012. The caloric restriction paradigm: implications for healthy human aging. Am. J. Hum. Biol. 24, 101–106. https://doi.org/10.1002/ajhb. 22243
[52] Colman, J.R., Anderson, R.M., Johnson, S.C., et al., 2009. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 325, 201–204. C
[53] Fontana, L., Nehme, J., Demaria, M., 2018. Caloric restriction and cellular senescence. Mech. Ageing Dev. 176, 19–23. https://doi.org/10.1016/j.mad.2018.10.005.
[54] Sanz, A., Caro, P., Ayala, V., Portero-Otín, M., Pamplona, R., Barja, G., 2006. Methionine restriction decreases mitochondrial oxygen radical generation and leak as well as oxidative damage to mitochondrial DNA and proteins. FASEB J. 20, 1064–1073. S
[55] Lambert, A.J., Buckingham, J.A., Boysen, H.M., Brand, M.D., 2010. Low complex I content explains the low hydrogen peroxide production rate of heart mitochondria from the long-lived pigeon, Columba livia. Aging Cell 9, 78–91. https://doi.org/10.1111/j. 1474-9726.2009.00538.x. (Epub 2009Nov 25.).
[56] Barja, G., 2007. Mitochondrial oxygen consumption and ROS production are independently modulated. Implications for aging studies. Rejuv Res 10, 215–223.
[57] Radak, Z., Torma, F., Berkes, I., et al., 2019. Exercise effects on physiological function during aging. Free Radic. Biol. Med. 132, 33–41. https://doi.org/10.1016/j. freeradbiomed.2018.10.444.
[58] Barja, G., 2013. Updating the mitochondrial free radical theory of aging: an integrated view, key aspects and confounding concepts. Antiox Redox Signaling 19, 1420–1445. https://doi.org/10.1089/ars.2012.5148.
[59] Barja, G., Cadenas, S., Rojas, C., Pérez-Campo, R., López-Torres, M., 1994. Low mitochondrial free radical production per unit O2 consumption can explain the simultaneous presence of high longevity and high metabolic rates in birds. Free Rad Res 21, 317–328.
[60] Recent developments in yeast aging. Kaeberlein M, Burtner CR, Kennedy BK PLoS Genet. 2007 May 25; 3(5):e84.
[61] Hayflick L. The future of ageing. Nature. 2000;408:37–39.
[62] Biological aging is no longer an unsolved problem. Hayflick L Ann N Y Acad Sci. 2007 Apr; 1100():1-13.
[63] Hayflick L. Entropy explains aging, genetic determinism explains longevity, and undefined terminology explains misunderstanding both. PLoS Genet. 2007 Dec;3(12):e220. doi: 10.1371/journal.pgen.0030220. PMID: 18085826; PMCID: PMC2134939.
[64]Cellular and molecular longevity pathways: the old and the new. Nikoletopoulou V, Kyriakakis E, Tavernarakis N. Trends Endocrinol Metab, 25(4):212-223, 31 Dec 2013
[65] Blagosklonny MV. Molecular damage in cancer: an argument for mTOR-driven aging. Aging (Albany NY) 2011; 3:1130 – 41; PMID: 22246147
[66] Blagosklonny MV. Damage-induced aging and perpetual motion. Cell Cycle. 2013;12(17):2709-2710. doi:10.4161/cc.26015MLABlagosklonny, Mikhail V. “Damage-induced aging and perpetual motion.” Cell cycle (Georgetown, Tex.) vol. 12,17 (2013): 2709-10. doi:10.4161/cc.26015APABlagosklonny M. V. (2013). Damage-induced aging and perpetual motion. Cell cycle (Georgetown, Tex.), 12(17), 2709–2710. https://doi.org/10.4161/cc.26015NLMBlagosklonny MV. Damage-induced aging and perpetual motion. Cell Cycle. 2013 Sep 1;12(17):2709-10. doi: 10.4161/cc.26015. Epub 2013 Aug 5. PMID: 23966155; PMCID: PMC3899178.
[67] Kenyon CJ. The genetics of ageing. Nature 2010; 464:504 – 12; http://dx.doi.org/10.1038/nature08980; PMID: 20336132
[68] Bartke A. Insulin and aging. Cell Cycle 2008; 7:3338 – 43; http://dx.doi.org/10.4161/cc.7.21.7012; PMID: 18948730
[69] Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW, Thomas EL, Kockel L. With TOR, less is more: a key role for the conserved nutrient‐sensing TOR pathway in aging. Cell Metab. 2010; 11:453‐465.
[70] Zoya N. Demidenko & Mikhail V Blagosklonny (2008) Growth stimulation leads to cellular senescence when the cell cycle is blocked, Cell Cycle, 7:21, 3355-3361, DOI: 10.4161/ cc.7.21.6919
[71] Growth stimulation leads to cellular senescence when the cell cycle is blocked. Demidenko ZN, Blagosklonny MV Cell Cycle. 2008 Nov 1; 7(21):3355-61.
[72] Alternative Perspectives on Aging in Caenorhabditis elegans: Reactive Oxygen Species or Hyperfunction? Gems D, de la Guardia Y Antioxid Redox Signal. 2013 Jul 20; 19(3):321-9.
[73] Patil CK, Mian IS, Campisi J. The thorny path linking cellular senescence to organismal aging. Mech Ageing Dev 2005; 126:1040-5.
[74] Blagosklonny MV. Rapamycin and quasi-programmed aging: four years later. Cell Cycle 2010; 9:1859 – 62; http://dx.doi.org/10.4161/cc.9.10.11872; PMID:
[75] Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature 2013; 493:338 – 45; http://dx.doi.org/10.1038/nature11861; PMID: 23325216
[76] Mikhail V Blagosklonny (2013) Aging is not programmed, Cell Cycle, 12:24, 3736-3742, DOI: 10.4161/cc.27188
[77] Alternative Perspectives on Aging in Caenorhabditis elegans: Reactive Oxygen Species or Hyperfunction? Gems D, de la Guardia Y Antioxid Redox Signal. 2013 Jul 20; 19(3):321-9.
[78] . Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011; 12:21‐35.